Irene KaplowI am an Assistant Professor in the Departments of Biological Sciences and Computational Biology at Carnegie Mellon University, where I am investigating the role of changes in transcriptional regulation in the evolution of vertebrate metabolic phenotypes and dissecting the sequence differences at regulatory element orthologs that have caused regulatory activity differences between species. If you are interested in being a Ph.D. student in my lab, apply to Carnegie Mellon University's Ph.D. Program in Biological Sciences or the Carnegie Mellon-University of Pittsburgh Ph.D. Program in Computational Biology. I received my B.S. in Mathematics with a minor in Biology from the Massachusetts Institute of Technology in 2010. There, I began my career as a computational biologist while doing research with Bonnie Berger. I next went to graduate school at Stanford University, where I received my Ph.D. in Computer Science in 2017. At Stanford, I worked in Hunter Fraser and Anshul Kundaje's labs to develop methods to analyze novel high-throughput sequencing datasets to better understand the roles of DNA methylation and Cys2-His2 zinc finger transcription factor binding in transcriptional regulation. I then worked as a Lane Postdoctoral Fellow in Andreas Pfenning's lab in the Computational Biology Department at Carnegie Mellon University, where I developed methods to identify regulatory elements whose regulatory activity differences between species are associated with the evolution of neurological phenotypes. After that, I worked as a research scientist in Charles Gersbach's lab in the Center for Advanced Genomics Technologies at Duke University, where I helped experimentally test the effects of manipulating a candidate myosin gene enhancer on myosin gene expression and cardiomyocyte development. |

|
Selected Publications |
|
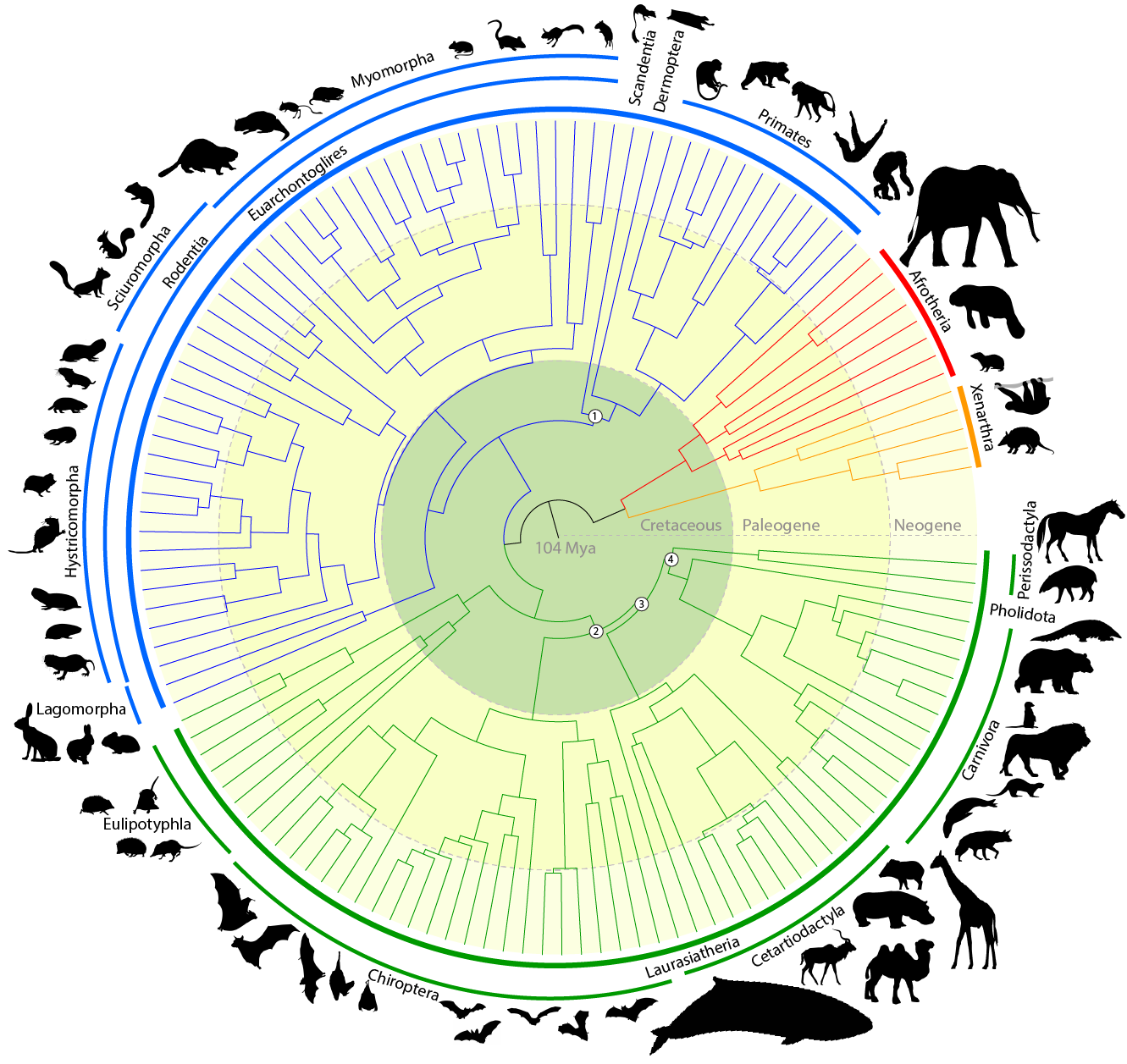
Evolutionary constraint and innovation across hundreds of placental mammals.Christmass MJ*, Kaplow IM*, Genereux DP, Dong MX, Hughes GM, Li X, Sullivan PF, Hindle AG, Andrews G, Armstrong JC, Bianchi M, Breit AM, Diekhans M, Fanter C, Foley NM, Goodman DB, Goodman L, Keough KC, Kirilenko B, Kowalczyk A, Lawless C, Lind AL, Meadows JRS, Moreira L, Redlich RW, Ryan L, Swofford R, Valenzuela A, Wagner F, Wallerman O, Brown AR, Damas J, Fan K, Gimshaw J, Johnson J, Kozyrev SV, Lawler AJ, Marinescu VD, Morril KM, Osmanski A, Paulat NS, Phan BN, Reilly SK, Schäffer DE, Steiner C, Supple MA, Wilder AP, Wirthlin ME, Xue JR, Zoonomia Consortium, Birren BW, Gazal S, Hubley RM, Koepfli KP, Marques-Bonet T, Meyer WK, Nweeia M, Sabeti PC, Shapiro B, Smit AFA, Springer M, Teeling E, Weng Z, Hiller M, Levesque DL, Lewin HA, Murphy WJ, Navarro A, Paten B, Pollard KS, Ray DA, Ruf I, Ryder OA, Pfenning AR, Lindblad-Toh K#, Karlsson EK# Science, 2023 PubMed link / code We compare genomes from hundreds of mammal species to explore features conserved by evolution and the origins of exceptional traits. |
|
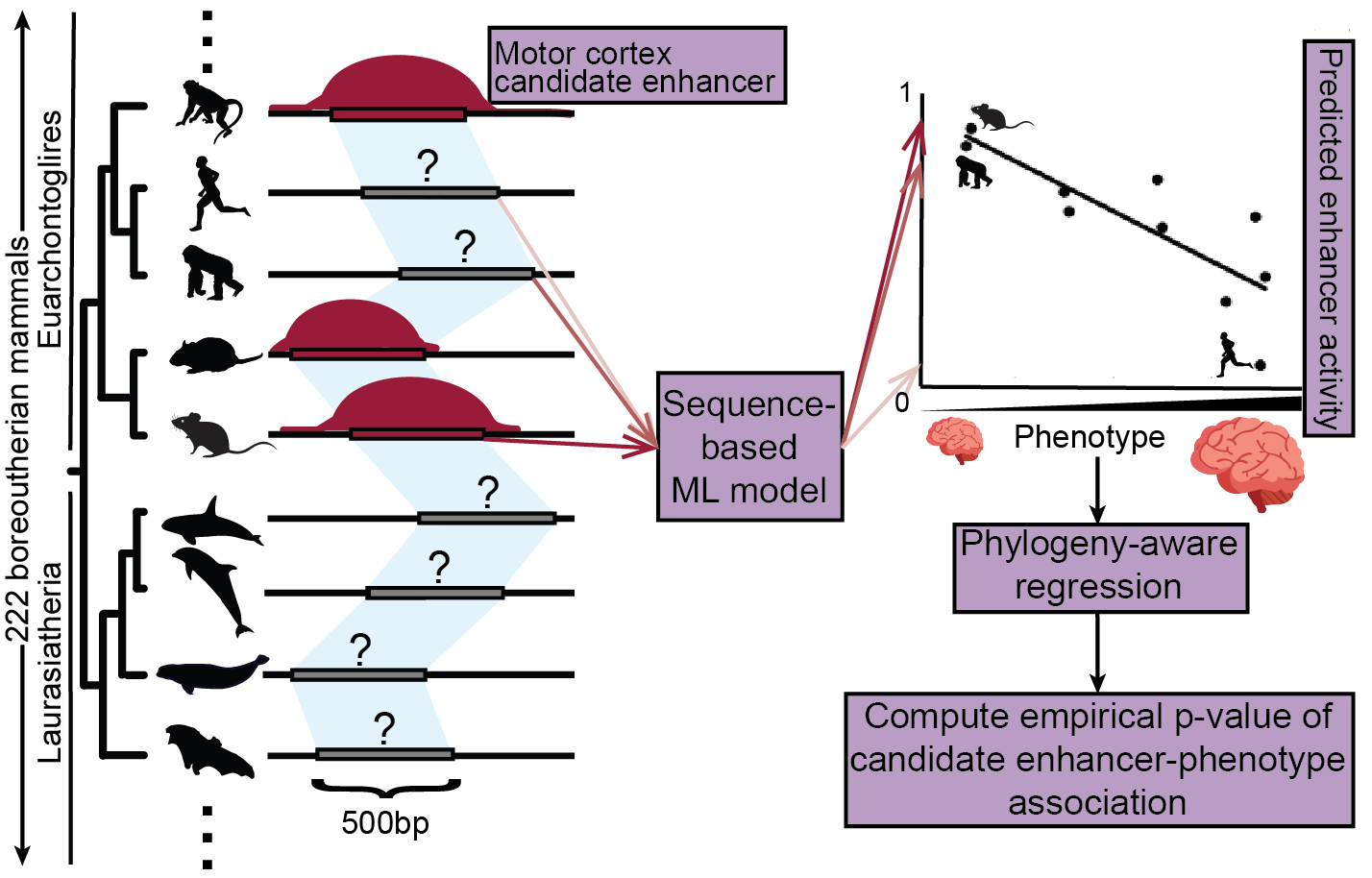
Relating enhancer genetic variation across mammals to complex phenotypes using machine learning.Kaplow IM*#, Lawler AJ*, Schäffer DE*, Srinivasan C, Sestili HH, Wirthlin ME, Phan BN, Prasad K, Brown AR, Zhang X, Foley K, Genereux DP, Zoonomia Consortium, Karlsson EK, Lindblad-Toh K, Meyer WK, Pfenning AR# Science, 2023 PubMed link / code A new machine learning-based approach associates enhancers with the evolution of brain size and behavior across mammals. |
|
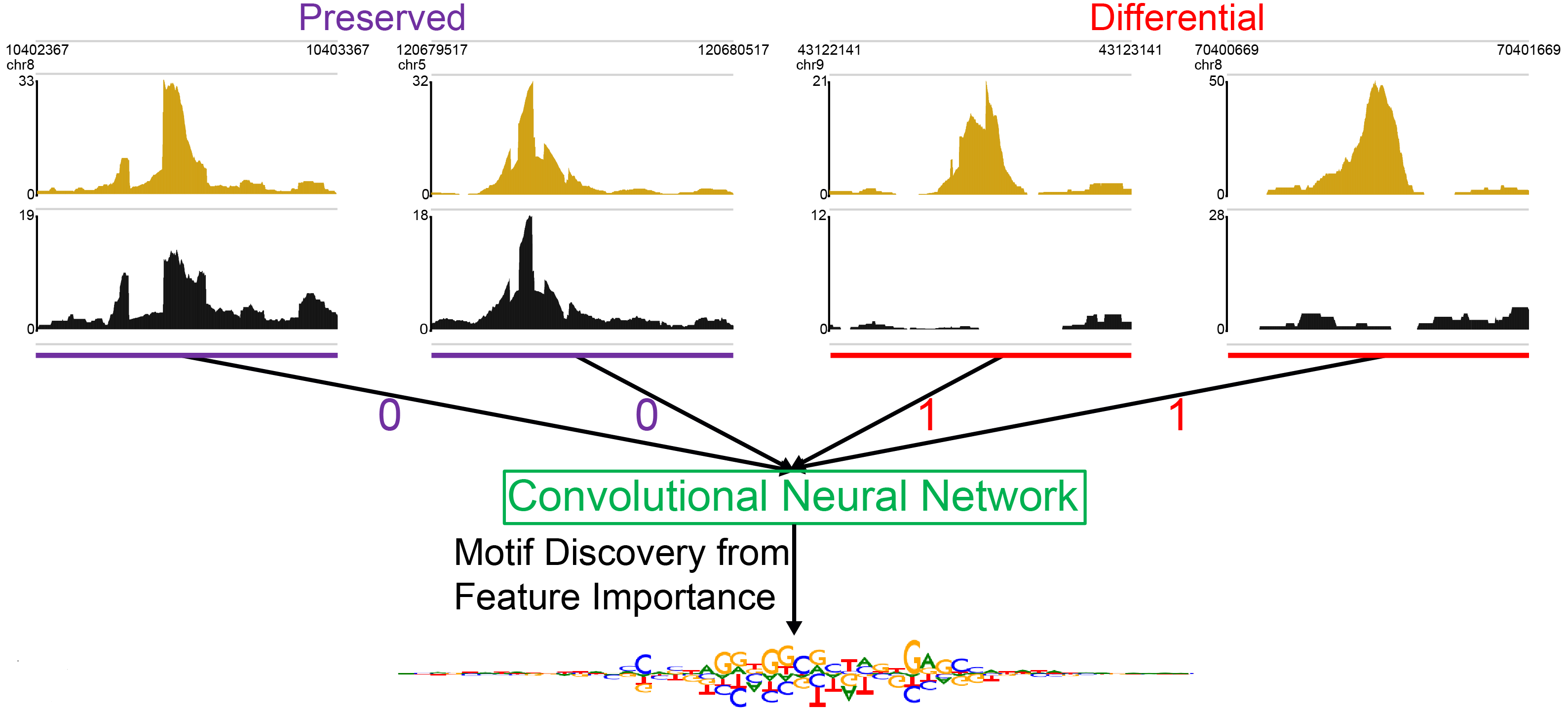
Neural network modeling of differential binding between wild-type and mutant CTCF reveals putative binding preferences for zinc fingers 1–2.Kaplow IM#, Banerjee A, Foo CS# BMC Genomics, 2022 PubMed link / code We developed a new approach to identifying the binding motifs of individual DNA binding domains of a transcription factor through analyzing chromatin immunoprecipitation sequencing experiments in which a single DNA binding domains is mutated and applied it to identify the known binding preferences of CTCF zinc fingers 3–11 as well as a putative GAG binding motif for zinc finger 1. |
|
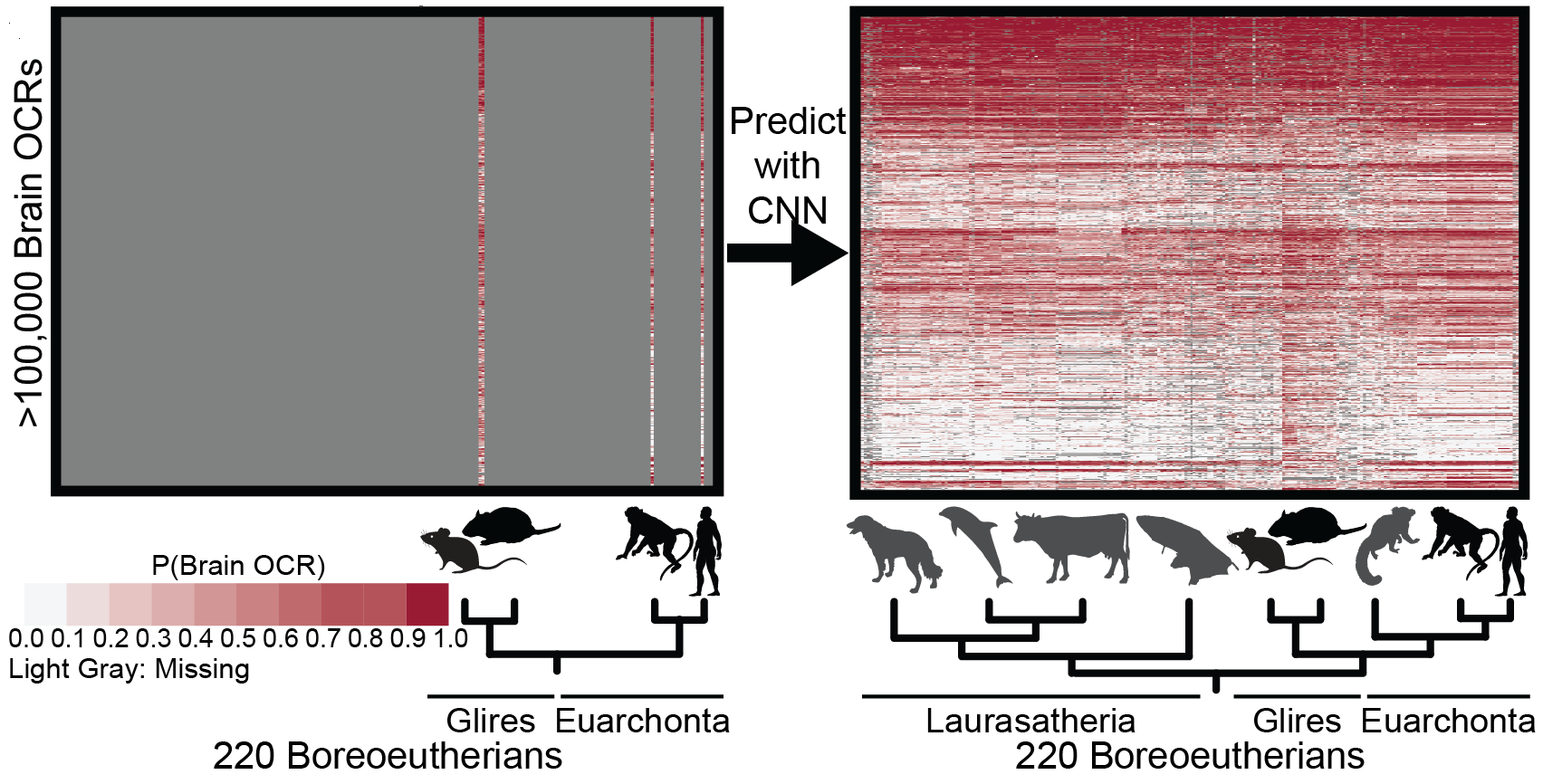
Inferring mammalian tissue-specific regulatory conservation by predicting tissue-specific differences in open chromatin.Kaplow IM#, Schäffer DE, Wirthlin ME, Lawler AJ, Brown AR, Kleyman M, Pfenning AR# BMC Genomics, 2022 PubMed link / code We provide a mechanism to annotate tissue-specific regulatory function across hundreds of genomes and to study enhancer evolution using predicted regulatory differences rather than nucleotide-level conservation measurements. |
|
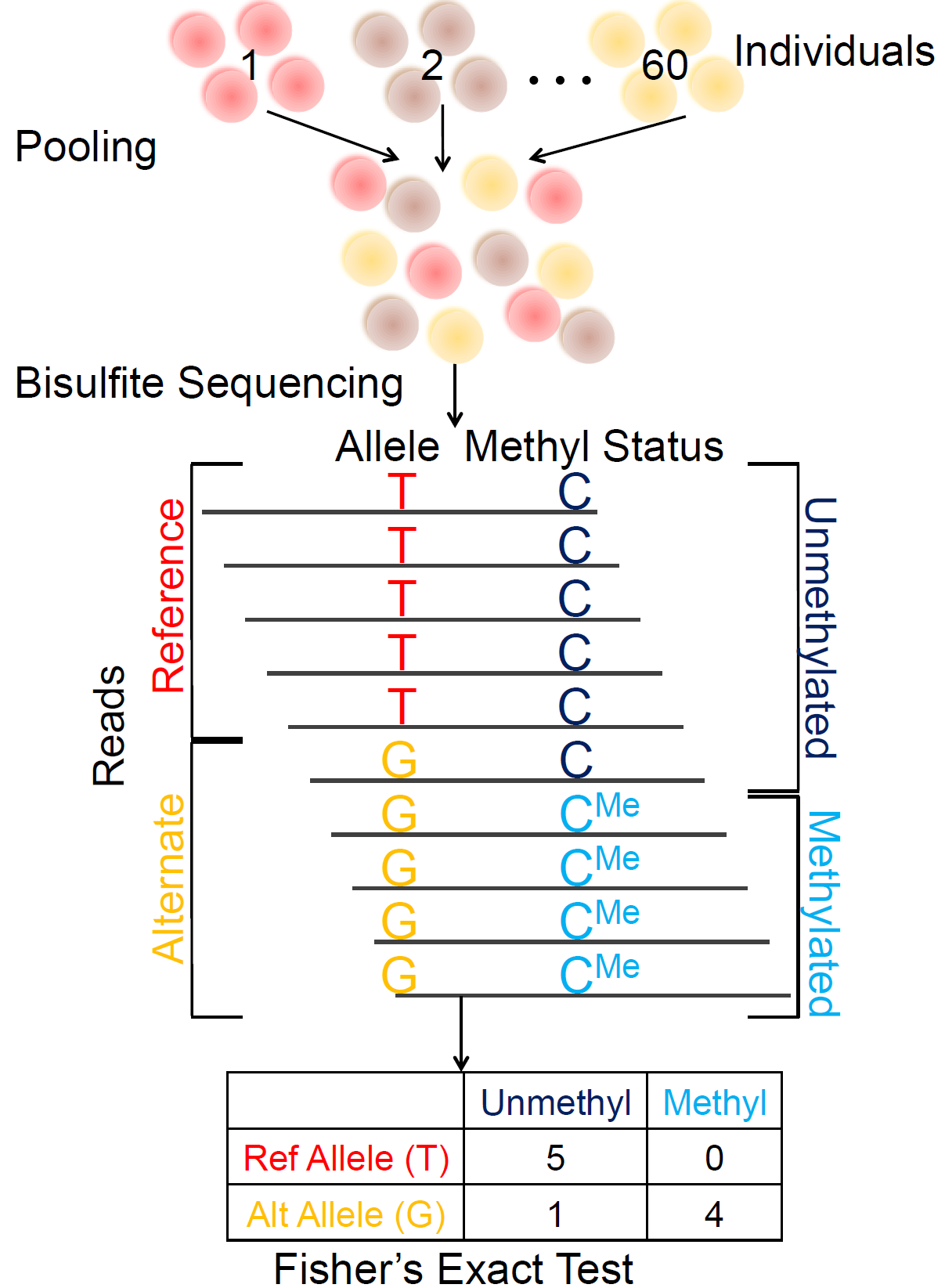
A pooling-based approach to mapping genetic variants associated with DNA methylation.Kaplow IM, MacIsaac JL*, Mah SM*, McEwen LM, Kobor MS, Fraser HB Genome Research, 2015 PubMed link We present a novel approach for identifying genetic variants associated with DNA meythlation in which bisulfite-treated DNA from many individuals is sequenced together in a single pool, resulting in a truly genome-wide DNA methylation map. |
Additional Publications |
A gene regulatory element modulates myosin expression and controls cardiomyocyte response to stress.Anglen T, Kaplow IM, Choi B, Dewars E, Perelli RM, Hagy KT, Tran D, Ramaker ME, Shah SH, Jung I, Landstrom AP, Karra R, Diao Y, Gersbach CA Genome Research, 2025 PubMed link / code We identify an enhancer that regulates Myh6 and show that activating the enhancer alters cardiac response to stress. |
An in vivo systemic massively parallel platform for deciphering animal tissue-specific regulatory function.Brown AR, Fox GA*, Kaplow IM*, Lawler AJ, Phan BN, Gadey L, Wirthlin ME, Ramamurthy E, May GE, Chen Z, Su Q, McManus CJ, van der Weerd R, Pfenning AR Frontiers in Genetics, 2025 PubMed link / code We present a new method for quantifying enhancer activity across tissues in vivo in which we deliver a massively parallel reporter assay directly into a mouse. |
Regression convolutional neural network models implicate peripheral immune regulatory variants in the predisposition to Alzheimer's disease.Ramamurthy E, Agarwal S, Toong N, Sestili H, Kaplow IM, Chen Z, Phan B, Pfenning AR PLoS Computational Biology, 2024 PubMed link / code We present a new way of thinking about training machine learning models to predict the effects of non-coding variants on regulatory element activity and implicate a role for immune cells in Alzheimer’s disease. |
Vocal learning-associated convergent evolution in mammalian proteins and regulatory elements.Wirthlin ME*, Schmid TA*, Elie JE*, Zhang X, Kowalczyk A, Redlich R, Shvareva VA, Rakuljic A, Ji MB, Bhat NS, Kaplow IM, Schäffer DE, Lawler AJ, Wang AZ, Phan BN, Annaldasula S, Brown AR, Lu T, Lim BK, Azim E, Zoonomia Consortium, Clark NL, Meyer WK, Pond SLK, Chikina M, Yartsev MM#, Pfenning AR# Science, 2024 PubMed link We identify an Egyptian fruit bat brain region involved in vocal learning and use it to find regulatory elements associated with vocal learning evolution in multiple parts of the mammalian phylogenetic tree. |
Machine learning sequence prioritization for cell type-specific enhancer design.Lawler AJ, Ramamurthy E, Brown AR, Shin N, Kim Y, Toong N, Kaplow IM, Wirthlin M, Zhang X, Fox G, Wade K, He J, Ozturk BE, Byrne LC, Stauffer WR, Fish KN, Pfenning AR Elife, 2022 PubMed link We present Specific Nuclear-Anchored Independent Labeling (SNAIL), which leverages machine learning and other computational approaches to identify DNA sequence features that confer cell type-specific gene activation and then makes a probe that drives an affinity purification-compatible reporter gene. |
Addiction-associated genetic variants implicate brain cell type- and region-specific cis-regulatory elements in addiction neurobiology.Srinivasan C*, Phan BN*, Lawler AJ, Ramamurthy E, Kleyman M, Brown AR, Kaplow IM, Wirthlin ME, Pfenning AR Journal of Neuroscience, 2021 PubMed link / code We combine statistical genetic and machine learning techniques to find that the predisposition to for nicotine, alcohol, and cannabis use behaviors can be partially explained by genetic variants in conserved regulatory elements within specific brain regions and neuronal subtypes of the reward system. |
Population-Specific Neuromodulation Prolongs Therapeutic Benefits of Deep Brain Stimulation.Spix TA, Nanivadekar S, Toong N, Kaplow IM, Isett BR, Goksen Y, Pfenning AR, Gittis AH Science, 2021 PubMed link / code We used synaptic differences to excite parvalbumin-expressing external globus pallidus (GPe) neurons and inhibit lim-homeobox-6–expressing GPe neurons simultaneously using brief bursts of electrical stimulation and found that, in dopamine-depleted (DD), circuit-inspired DBS provided long-lasting therapeutic benefits that far exceeded those induced by conventional DBS. |
HALPER facilitates the identification of regulatory element orthologs across species.Zhang X*, Kaplow IM*, Wirthlin M, Park TY, Pfenning AR Bioinformatics, 2020 PubMed link / code We present halLiftover Post-processing for the Evolution of Regulatory Elements (HALPER), a tool for constructing contiguous regulatory element orthologs from the outputs of the halLiftover tool for mapping genomic regions across Cactus multi-species alignments. |
Mitigation of off-target toxicity in CRISPR-Cas9 screens for essential non-coding elements.Tycko J*, Wainberg M*, Marinov G*, Ursu O, Hess G, Ego B, Aradhana A, Li A, Truong A, Trevino A, Spees K, Yao D, Kaplow IM, Greenside P, Morgens D, Phanstiel D, Snyder M, Bintu L, Greenleaf W#, Kundaje A#, Bassik M# Nature Communications, 2019 PubMed link / code We performed a genome-wide non-coding screen for essential CTCF sites in chromatin loop anchors in the K562 leukemia cell line and discovered that the dominant source of signal in our screen was not due to deregulated gene expression but was instead consistent with CRISPR-Cas9 off-target activity causing reductions in cell fitness, despite filtering the single guide RNAs (sgRNAs) to have no perfect or 1-mismatch off-target sites. |
Fine-mapping cis-regulatory variants in diverse human populations.Tehranchi A, Hie B, Dacre M, Kaplow IM, Pettie K, Combs P, Fraser HB Elife, 2019 PubMed link / code To better understand human cis-regulatory variation, we mapped quantitative trait loci for chromatin accessibility (caQTLs)-a key step in cis-regulation-in 1000 individuals from 10 diverse populations and found that many caQTLs that affect the expression of distal genes also alter the landscape of long-range chromosomal interactions, suggesting a mechanism for long-range expression QTLs. |
Two DNA-encoded strategies for increasing expression with opposing effects on promoter dynamics and transcriptional noise.Dadiani M, van Dijk D, Segal B, Field Y, Ben-Artzi G, Raveh-Sadka T, Levo M, Kaplow IM, Weinberger A, Segal E Genome Research, 2013 PubMed link We used single-cell time-lapse microscopy to compare the effect on transcriptional dynamics of two distinct types of sequence changes in the promoter that can each increase the mean expression of a cell population by similar amounts but through different mechanisms and showed that increasing expression by strengthening a transcription factor binding site results in slower promoter dynamics and higher noise as compared with increasing expression by adding nucleosome-disfavoring sequences. |
RNAiCut: Automated detection of significant genes from functional genomic screens.Kaplow IM*, Singh R*, Friedman A, Bakal C, Perrimon N#, Berger B# Nature Methods, 2009 PubMed link We built a fully automated system, RNAiCut, that objectively identifies score thresholds from RNA interference (RNAi) data by introducing the use of the connectivity of subgraphs of protein-protein interaction (PPI) networks. |
A classifier-based approach to identify genetic similarities between diseases.Schaub MA, Kaplow IM, Sirota M, Do CB, Butte AJ, Batzoglou S Bioinformatics, 2009 PubMed link We introduce a novel methodology that identifies similarities between diseases in which we train a classifier on single nucleotide polymorphism (SNP) data that distinguishes between individuals that have one disease and a set of control individuals and then use the classifer to classify individuals with a second disease and apply the method to reveal the known genetic similarity between type 1 diabetes and rheumatoid arthritis and a new putative similarity between bipolar disease and hypertension. |
|
Design and source code from Leonid Keselman's website |